GODDESS / GRASS
is a software to simulate 13C NMR spectra of oligo- and polysaccharides, and to predict structure from spectra.Glycan NMR spectra simulation
Glycan-Optimized Database-Driven Empirical Spectra Simulation (GODDESS) accumulates two empirical approaches to the 13C NMR spectra simulation:1. Incremental scheme (13C only) composes subspectra of monosaccharides and other residues from dedicated database of mono-, di- and trimeric fragments, theoretical substitution effects and steric strain effects. It was initially deployed as Biopolymer Structure Elucidation (BIOPSEL) software in 2001. Since then it has been improved to accomodate greater variety of residues and structural features, including non-carbohydrate constituents, and got a web-interface. In 2013 we proved that application of this scheme on water solutions of natural glycans outperformed quantum-mechanical NMR calculations in large basis sets, such as B3LYP/6-311G++(2d,2p) or PBE/PBE both in accuracy and speed [ref]. Click here for more details on the project web-site.
2. Statistical scheme (13C and 1H) adopts HOSE idea at the level of residues and utilizes heuristic algorithm of structure generalization tuned for carbohydrates. This approach does not need dedicated databases but uses a large and regularly updated database (CSDB, >4000 spectra). It generalises structural surrounding of the atom under prediction until enough structurally-similar fragments are found in the database and averages found chemical shifts with outlier removal. Depending on the generalization type, stereochemistry and distance from the simulation site, a weight factor is assigned to every generalization act. Minimal total weight is a criterion of finding out the best generalization pathway. In 2015 we achieved average simulation accuracy on bioglycans and glycoconjugates as 0.86 ppm per 13C resonance and 0.07 ppm per 1H resonance [ref]. Click here for more details on the project web-site.
3. Both approaches report trustwothiness and/or accuracy of every atom simulation. Based on these values and dataset size and dispersion, a hybrid scheme (13C only) combines the results from the two approaches using flexible scale factors. Click here for more details on the project web-site.
Database-driven schemes allow tracking of assumptions, generalizations and chemical shifts down to original published data. Both approaches are available as features of Carbohydrate Structure Database. To enter a glycan structure and run simulation, click Extras/Predict NMR in the CSDB left menu or use a direct link: NMR simulation. Output includes one- and two-dimensional simulated spectra and signal assignment tables, exemplified below:
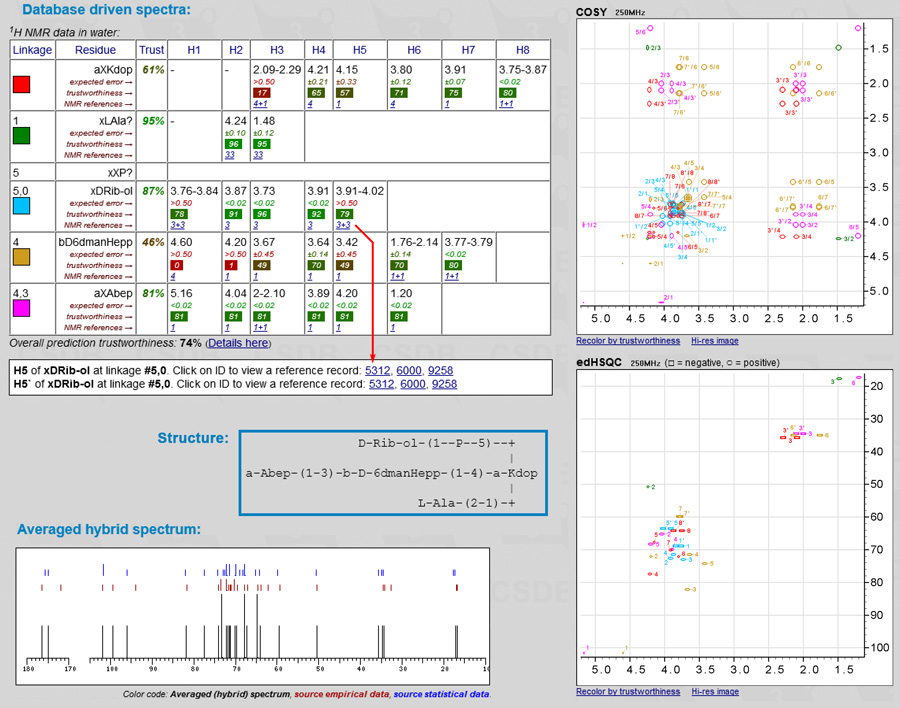
Currently, the following experiments can be schematically visualized at any spectrometer frequency (plain or assigned): 1D 13C, COSY, COSY RCT, COSY DQF, TOCSY, edHSQC, HSQC-TOCSY and HMBC. Accuracy of predictions on two typical bioglycan structures is shown in the figures:
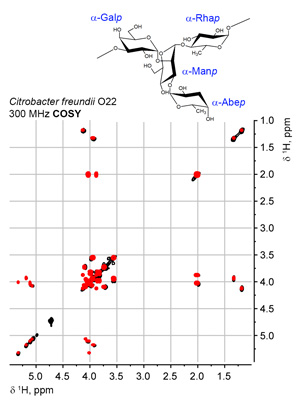
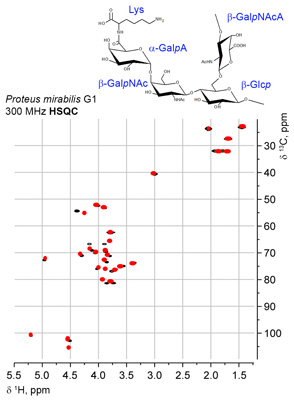
NMR-based structure ranking
Generation, Ranking and Assignment of Saccharide Structures (GRASS) is a structural iterator, which generates all possible saccharide-containing oligo- and polymers within the specified structural constraints. The only mandatory constraint is a number of residues per oligomer or polymer repeat unit, but the accuracy can be improved by other contraints, such as number of CH2 carbons, number of β-sugars, methylation analysis data, GC monomeric composition, absolute, anomeric or ringsize configurations, partial sequence data etc. A fast empirical 13C NMR spectrum simulator is called for every structure, and ≤500 best matches are refined by the slower but more accurate statistical simulator. The algorithm is tolerant to missing or extra signals in the inputed experimental spectrum. Structural hypotheses are ranked accordingly to the similarity between experimental and simulated spectra:
| INPUT: |
PREDICTION:| OUTPUT: |
|
